В настоящее время, характеристики низкоразмерных систем, которые сложно и/или дорого исследовать экспериментально, благодаря развитию современной вычислительной техники и методов решения больших систем уравнений, можно получить с высокой точностью при расчете свойств веществ из первых принципов.
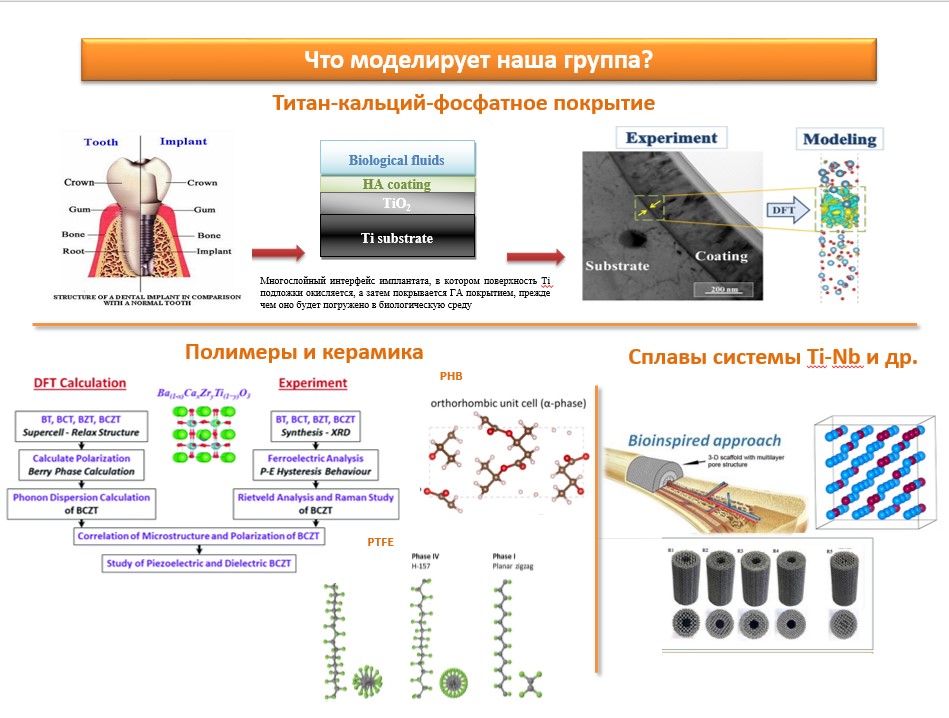
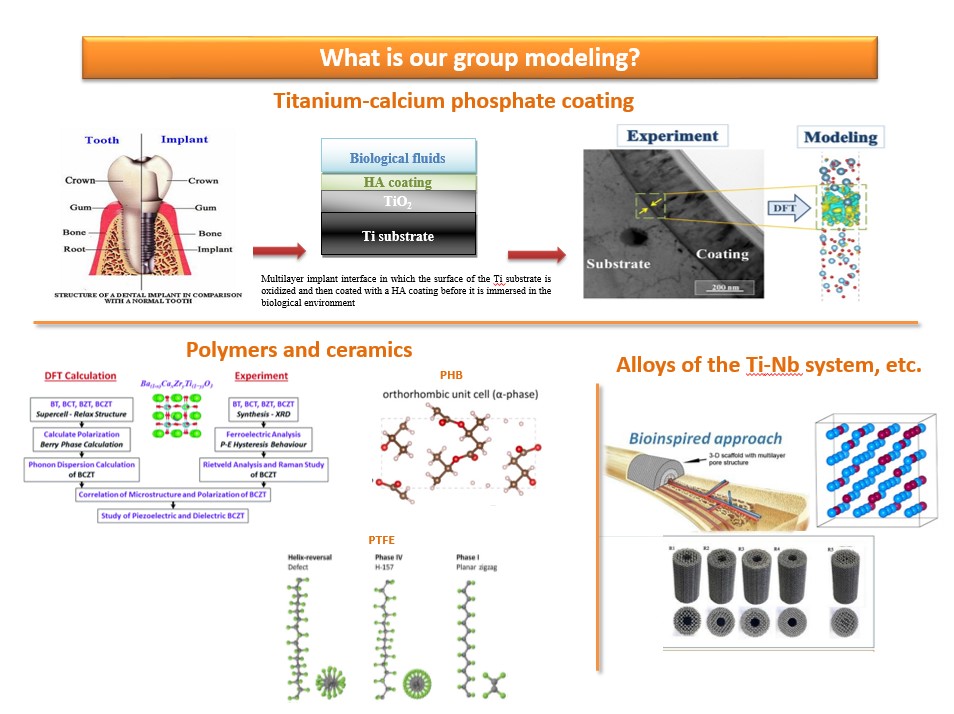
В последние годы расчетный дизайн материалов, основанный на первых принципах квантово-механических методов, стал важным экономически эффективным инструментом для прогнозирования свойств веществ. В нашей работе мы используем компьютерное моделирование в VASP для исследования различных свойств кальций-фосфатных покрытий, сформированных на титане, Ti-Nb сплавов, а также полимерных материалов, применяемых в биомедицине. Использование теоретических прогнозов свойств материалов способны помочь снизить затраченные усилия, обычно возникающие при экспериментальном изучении веществ с помощью широкого комплекса методов (Рисунок 1). Основные темы, представленных на рисунке 1 направлений исследований сформулированы ниже:
· Механизмы межатомного взаимодействия на границе раздела титан-кальций-фосфатное покрытие: первопринципное исследование;
· Оценка закономерностей влияния микроструктуры на электрические свойства пьезоэлектрической керамики BCZT;
· Структурные, электронные, упругие и пьезоэлектрические свойства поли(3-гидроксибутирата) (ПГБ): первопринципеное исследование;
· Численное моделирование термодинамических и физико-механических свойств сплава системы Ti-Nb, Ti-Nb-Zr-Ta.
Программный пакет VASP представляет собой вычислительный комплекс для проведения компьютерного моделирования многоэлектронных систем атомных масштабов, применяемый для численного решения задач квантово-механической молекулярной динамики и расчётов, связанных с электронной структурой. В данном программном пакете применяется теория функционала плотности, которая является одним из самых эффективных и точных методов исследования электронных свойств низкоразмерных систем и наноструктур.